
医药GMP无尘车间空气洁净度等级要求是多少?“药品生产质量管理规范”(GMP)中规定:药品生产的无尘车间内的生产环境参数如:温度和相对湿度以及压差等均是由生产工艺决定的,一般温度为18℃~24℃,相对湿度为45%~65%。

无菌药品的生产所需的洁净区可以分为四个级别:
A 级:高风险操作区,如灌装区、放置胶塞桶和与无菌制剂直接接触的敞口包装容器的区域及无菌装配或连接操作的区域,应当用单向流操作台(罩)维持该区的环境状态。单向流系统在其工作区域必须均匀送风,风速为0.36-0.54m/s(指导值)。应当有数据证明单向流的状态并经过验证。在密闭的隔离操作器或手套箱内,可使用较低的风速。
B 级:指无菌配制和灌装等高风险操作A 级洁净区所处的背景区域。
C 级和D 级:指无菌药品生产过程中重要程度较低操作步骤的洁净区。
以上各级别医药无尘车间空气悬浮粒子的标准与ISO14644-1中洁净度等级(以≥0.5μm和≥5μm的悬浮粒子为限度标准)的关系如下:
洁净区空气悬浮粒子的标准(用尘埃粒子计数器检测)
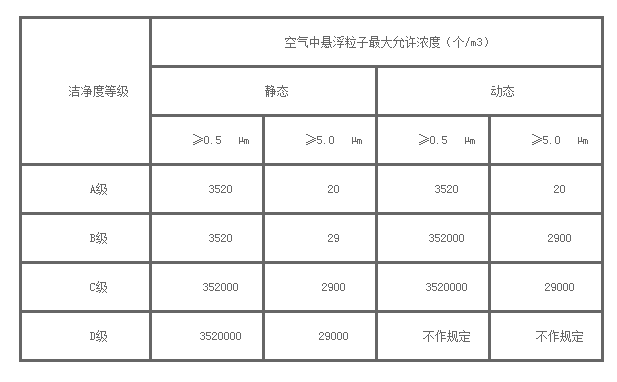
洁净区微生物监测的动态等级标准(可用浮游菌采样器检测)

